.png)

The term "Software as a Medical Device" is defined by the International Medical Device Regulators Forum (IMDRF) as "software intended to be used for one or more medical purposes that perform these purposes without being part of a hardware medical device." Software clearly needs hardware to run on; in this case it needs a general purpose platform, such as a PC, mobile device, or server. The software can process the output (ex. radiology images) from a medical device, as long as it is not part of the device (i.e. necessary to achieve the device's intended purpose). SaMD cannot drive a medical device - that software is considered part of the device. But it can interface with the device.

Overall, SaMD have the same regulatory requirements as traditional medical devices, except without electro-mechanical aspects and with a few additional aspects that are specific to SaMD. The sections below summarize the key elements of the SaMD submission.
The definition statement is where you, the manufacturer, state the intended Medical Purpose of your SaMD. Is it to Treat or Diagnose? Is it driving clinical management or informing clinical management? What is the significance of the information that the SaMD provides to a healthcare decision? What is the state of the situation/condition - Critical? Serious? Non-serious?
From this information, the risk is categorized into four levels, with category IV being highest impact, and I being lowest. The chart below describes how how these categories are established:
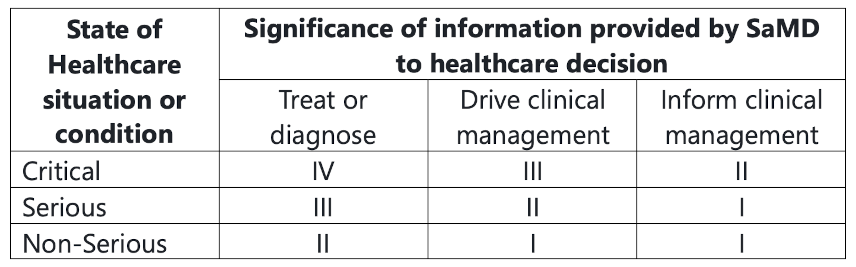
This categorization establishes the risks associated with the SaMD and the scrutiny of the design controls and clinical evaluation in the submission.
The risk management process follows ISO 14971 with the software specifics defined in IEC 62304. As for all software for medical devices, identification of software that could contribute to a hazardous situation must be analyzed. What would a bug in the critical part of the software potentially cause? Could the software be misused in a way that would be dangerous? What would be the consequences of unexpected behavior of third-party libraries (Software of Unknown Provenance/SOUP)? These are the types of questions the analysis should be asking to determine the risks. Risk control measures can be applied to reduce the risk, and proof of verification of these measures needs to be provided.
Cybersecurity is a big part of the SaMD risk management because of the connectivity generally provided and the general-purpose nature of the platforms on which they run. The FDA has a comprehensive draft (at time of writing) premarket guidance for the "recommendations" of the agency around the Cybersecurity. It has been our experience that these are requirements and are indeed a major focus of the FDA in submissions. With the number of hospitals getting attacked, it is no wonder that the FDA wants to make sure that the devices are not vectors of attacks, and SaMD is no exception. Cybersecurity threat modeling and risk analysis, as well as thorough descriptions of the design controls used to address the risks, are required.
Cloud software cybersecurity can and should be certified for any vendor used. SOC2, HiTrust, or ISO 27001 certification ensures the cloud security design and processes are in place to safeguard information.
For medical devices, quality management system requirements are generally pretty clear to manufacturers. But with the introduction of SaMD and its closeness to general non-regulated health apps, many SaMD providers don't realize that the Quality Management Principles must be applied. IEC 13485 provides the regulatory requirements of medical device quality management systems, and its implications to the organization and lifecycle processes must be followed. ISO 13485 certification of your software provider ensures that indeed the principals and controls are in place.
Clinical Evaluation includes the proposed association between a SaMD output and a Clinical Condition. What evidence, research, literature, data analysis, and clinical trials support the assertions? This is followed up by the proof of performance, showing the accuracy, reliability, and precision of the results, along with the clinical validation displaying that the output data achieves the intended purpose in the context of clinical care.
Evidence demonstrating a clinical association, with analytical and clinical validation must be provided. We have seen startups swayed by claims by providers (ex: blood pressure measurements using a mobile video camera), but without supporting data to prove the claim, it will not get through the regulators.
SaMD often provides a pathway for continuous post-market improvements, leveraging real-work performance data. Changes in the clinical evaluation or definition may necessitate a new 510k.
Regulators are grappling with AI/ML. To ensure patient safety, typically only "locked" algorithms have been accepted - learning stops before final test and production. But the regulators recognize that post-market adaptive/continual learning is incredibly powerful and has the ability to be transformative. At the time of writing, a total product lifecycle (TPLC) approach is being considered by the FDA. After publishing a framework for review, they recently drafted an Action Plan to address Artificial Intelligence and Machine Learning. They plan to update their proposed framework and issue guidance on Predetermined Change Control - how the machine learning algorithm will change by learning, while remaining safe and effective. Harmonization of Good Machine Learning Practice (GMLP) will be encouraged, describing the set of best practices. Efforts by regulatory science to develop methodologies for improvements that can identify and eliminate bias will be supported. And overall, a Patient-centered approach will be necessitated. Stay tuned.

